使用OPLSAA构建模型
# 使用OPLSAA构建模型
moltemplate不仅可以通过键接信息构建其余几何信息,还可以通过预置的力场文件自动分配电荷、键角种类和势函数参数。
# 简介
OPLS是optimized potentials for liquid simulations,从其全称可知其适用范围,要是液体体系。该力场由Jorgensen团队开发,主要适用于多肽、蛋白、核酸、有机溶剂等液体体系,一般和TIP3P或TIP4P的水模型搭配适用。
为了便于移植,这个力场的势函数适用了常见的势函数,如下:
with the combining rules:
该力场在计算非间相互作用作用时,完全排除了1-2, 1-3对作用,而对于1-4对作用,采用了保留50%的策略。其他对相互作用,完全考虑;且对于LJ势参数,采用了Lorentz-Berthelot混合法则(几何平均,lammps的geometric)进行计算。
OPLS包含两套力场,OPLS-AA(全原子力场)和OPLS-UA(联合原子力场,不考虑氢原子)。LAMMPS选择如下命令来处理上面的相互作用:
units real
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style opls
improper_style harmonic
pair_style lj/cut/coul/long 10.0 10.0
pair_modify mix geometric
special_bonds lj/coul 0.0 0.0 0.5
kspace_style pppm 0.0001
2
3
4
5
6
7
8
9
10
力场文件从这里 (opens new window)下载。
#atom type class name group atomic number mass idk
atom 1 1 F "Fluoride -CH2-F (UA)" 9 18.998 1
2
3
接下来的参数部分均以class数值为准,不同原子间采用几何平均混合。
使用moltemplate配合OPLSAA建模,为了自动分配拓扑链接的种类,请使用已经写好的oplsaa.lt (opens new window)格式力场文件,或者在moltemplate/force_fields中自取
# 单独用OPLSAA力场
我们以一个简单的乙烷分子为例,来考虑如何使用moltemplate构建LAMMPS输入数据。
0.1 H H
\ /
0 H — C —— C — H
/ \
-0.1 H H
0 0.1|0.2 0.3
0.15
2
3
4
5
6
7
8
9
使用moltemplate的思路通常是从下到上考虑,即将大分子拆分成可复用的最小单元,然后再进行组合。因此,我们可以选择两个CH3-作为基本单元,再旋转、平移、链接乙烷。
首先构建CH3-
# CH3.lt
import "oplsaa.lt"
CH3 inherits OPLSAA {
write("Data Atoms"){
# AtomID MolID AtomType charge X Y Z
# $atom: $mol:. @atom:P4 0 0 0 0
# AtomID 按顺序赋予编号即可,不能重复
# MolID .令moltemplate自动按分子赋值
# @atom 从oplsaa.lt中charge部分查找种类编号,可以和Mass部分交叉验证
# charge 全部置零,生成时会按照力场自动替换
# 即便建模时非常生硬,在lammps 的minimize之后也可以迅速达到正常构型
$atom:1 $mol:. @atom:85 0.0 0 0 0 # "Alkane H-C"
$atom:2 $mol:. @atom:85 0.0 0 0.1 0
$atom:3 $mol:. @atom:85 0.0 0 -0.1 0
$atom:4 $mol:. @atom:80 0.0 0.1 0 0 # "Alkane CH3-"
}
write("Data Bond List"){
# 不需要手动输入bond type,会根据力场中的原子配对自动补全
# 同理,生成angle、dihedral、improper时也会自动补全他们的类型
# 这是这个软件最强大的地方
$bond:1 $atom:1 $atom:4
$bond:2 $atom:2 $atom:4
$bond:3 $atom:3 $atom:4
}
}
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
Ok,我们拿到了一个CH3-,现在只需要把他旋转,然后两个连起来:
# ethane.lt
import "CH3.lt"
import "oplsaa.lt"
Ethane inherits OPLSAA {
CH3L = new CH3
# 以Z轴为轴(0, 0, 1),以(0.15, 0, 0)为原点旋转180°
CH3R = new CH3.rot(180, 0, 0, 1, 0.15, 0, 0)
write("Data Bond List"){
# 选择CH3L和CH3R中id为4的原子连接起来,
# moltemplate会自动推断这两个原子的类型以选择bond type
$bond:1 $atom:CH3L/4 $atom:CH3R/4
}
}
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
最后在system.lt中new一个新分子:
import "alkene.lt"
Alkene = new Alkene
# How big is the simulation box?
write_once("Data Boundary") {
0 50.0 xlo xhi
0 50.0 ylo yhi
0 50.0 zlo zhi
}
2
3
4
5
6
7
8
9
10
11
12
13
14
敲上命令然后回车:
moltemplate.sh system.lt
叮咚,我们就得到了乙烷的数据文件:
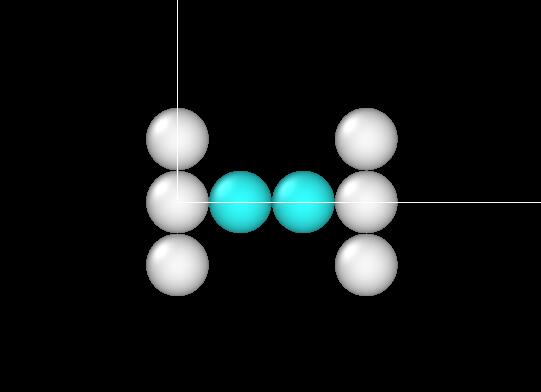
# 联合TIP4P力场
TIP4P是指水分子由四个位点组成,H2O三个,加一个没有质量只有电荷的虚原子(dummy atom)。可以从oplsaa.lt中看到,type 65-67 是TIP4P水的类型,其中M是虚原子。
OPLSAA联合TIP4P应用在LAMMPS会有一定的不兼容。具体问题是,如果显式地引入一个虚原子,转入npt阶段会抛出错误out of range - cannot compute pppm。现在大多数人都是修改一下,使用隐式的虚原子
moltemplate/examples/all_atom中有大量的使用预置力场的例子,可以交叉参考帮助理解
再次说明,小分子内的人工(生硬)构象再一次minimize之后就可以基本达到平衡,只需要保证拓扑链接准确即可。
# 隐式的虚原子
首先构建一个水分子,不需要引入虚原子M
# OPLSAA配合TIP4P 隐式式dummy_atom
# 注意OHH的顺序不能颠倒
import "oplsaa.lt"
Water inherits OPLSAA {
write("Data Atoms") {
$atom:o $mol:. @atom:65 -0.8476 0.0000000 0.000000 0.00000
$atom:h1 $mol:. @atom:66 0.4238 0.8164904 0.5773590 0.00000
$atom:h2 $mol:. @atom:66 0.4238 -0.8164904 0.5773590 0.00000
}
write("Data Bond List") {
$bond:1 $atom:o $atom:h1
$bond:2 $atom:o $atom:h2
}
}
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
先通过moltemplate.sh直接生成系统,然后再执行cleanup_moltemplate.sh清理到冗杂的力场信息。
然后我们修改system.in.init,将pair_style lj/cut/coul/long改为pair_style lj/cut/tip4p/long,kspace_style pppm 改成pppm/tip4p,参数不变 。请参考手册
(opens new window),tip4p/long是在coul/long的基础上增加了添加隐式虚原子的功能。此时我们需要在末尾cutoff之前给出描述虚原子位置的参数
lj/cut/tip4p/long args = otype htype btype atype qdist cutoff (cutoff2)
otype,htype = TIP4P H 和 O的atom type
btype,atype = bond type 和 angle type
qdist = 虚原子M与O的距离 (distance units)
cutoff = 全局LJ截断距离
cutoff2 = 库仑力的全局截断距离
2
3
4
5
6
7
同时我们还需要水分子的原子保持OHH的顺序,例如第500个原子是一个水的O,那501和502则是水的两个H,这样才能保证正确插入虚原子。
完成这一步以后,将system.in.settings中set charge命令删除,在system.in.charges中将O的电荷由0.0改为-1.04(也就是OPLSAA中M的电荷)。
最后使用fix shake锁住OH键。注意这个命令不能同minimize和fix nve/limit等积分器同用。