moltemplate
# moltemplate
# 概述
长久以来,lammps的数据文件构建就是一个大问题。一方面,LAMMPS的数据格式比较特殊,很多软件不能直接导出; 另一方面小分子到大分子的变换,大分子在系统中的排布和大分子中的拓扑结构都需要牵扯很多的精力。经过长时间的试错,终于找到一个比较合理且简约的技术栈,可以直接生成lammps所需的in文件和data文件。
如图所示,整个技术栈分为:
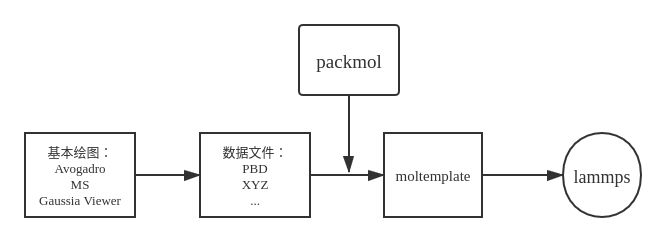
- 基本绘图:绘制基本的单元,如高分子结构单元,单个水分子或者甲烷分子等;
- 数据文件:导出成常见格式的数据文件,如PDB,XYZ;
- 空间排布(可选):packmol可以自动地根据所设置的限制向系统中填放单元。比如想向C60笼中填放原子,只需要一行命令将允许范围限制在球内即可;
- 生成工具:moltemplate可以操作已有的数据文件,在系统中复制,变换和随机,同时根据已有的拓扑结构补充生成键角二面角等信息,最后生成lammps所需的in文件和data文件。
# 文件结构
moltemplate本身具有导入(import)和继承(inherit)功能,因此我们可以尽量地把文件区分按照功能区分开。当然文件名都是可以自定义的,这里只是介绍一个思路,告诉各位应该怎样上手。
第一部分是forcefield.lt 这个文件中储存着lammps的系统和力场参数。
# -- ForceField -- #
ForceField{
write_once("In Init"){
units lj
boundary p p p
atom_style full
pair_style lj/cut 10.5
bond_style hybrid fene harmonic
angle_style harmonic
}
write_once("Data Boundary") {
0 100.0 xlo xhi
0 100.0 ylo yhi
0 100.0 zlo zhi
}
write_once("Data Masses"){
@atom:M 1
@atom:G 1
@atom:R 1
}
write_once("In Settings"){
pair_coeff @atom:M @atom:M 1 1 2.5
pair_coeff @atom:M @atom:G 1 1 2.5
pair_coeff @atom:M @atom:R 1 1 2.5
pair_coeff @atom:G @atom:G 1 1 2.5
pair_coeff @atom:G @atom:R 1 1 2.5
pair_coeff @atom:R @atom:R 1 1 2.5
}
write_once("In Settings"){
bond_coeff @bond:MM fene 30 1.5
bond_coeff @bond:GR fene 30 1.5
bond_coeff @bond:RR harmonic 1000 0.66
}
write_once("In Settings"){
angle_coeff @angle:RRR 200 180
}
# write_once("Data Dihedrals By Type"){
# @dihedral: @atom:
# }
# write_once("Data Angles By Type"){
# @angle: @atom:
# }
}
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
第二部分是基本单元数据 这里面存储这一个片段的信息
# -- file matrix.lt --
import "forcefield.lt"
Matrix inherits ForceField{
write("Data Atoms"){
$atom:1 $mol:. @atom:M 0 1 1 1
$atom:2 $mol:. @atom:M 0 2 1 1
$atom:3 $mol:. @atom:M 0 3 1 1
$atom:4 $mol:. @atom:M 0 4 1 1
$atom:5 $mol:. @atom:M 0 5 1 1
}
write("Data Bonds"){
$bond:1 @bond:MM $atom:1 $atom:2
$bond:2 @bond:MM $atom:2 $atom:3
$bond:3 @bond:MM $atom:3 $atom:4
$bond:4 @bond:MM $atom:4 $atom:5
}
}
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
第三部分是操作部分 在这个文件中我们可以对基本单元进行操作,同时也是程序执行的入口
# -- file system.lt -- #
import "forcefield.lt"
import "matrix.lt"
m1 = new Matrix [3].move(0,0,3)
2
3
4
5
6
在最简单的情况下,我们仅需要这三个文件就可以描述一个系统的初始化状态,然后执行命令行使用moltemplate生成实际文件:
moltemplate.sh system.lt
得到:
system.data
system.in
system.in.init
system.in.settings
# 如果出了bug还会出现生成树output_ttree
2
3
4
5
# 语法教程
# 1.1 write_once() 和 write()
write_once("file_name"){
text_content
}
2
3
每出现一次write_once(),就会新生成一个文件,整个程序执行过程中此命令仅仅执行一次。注意,In 和 Data 开头是系统保留关键词,将会被合并到.in和.data文件中,而特别离谱的才会被单独创立。比如,In Init就是个保留文件名,如果你输入In init就会被强制要求改成大写。接下来会有专门的专题来介绍保留文件名的特殊之处。还有一些文件名会被合并,比如Data Mass、Data boundary。write()则是可以在一个文本块内出现多次、可以被重复执行的单元,每new一次就会被写一次。
# import 和 inherits
import 关键字要求系统去查找需要引用的文件的位置
inherits 表示本结构单元中的变量名称(比如bond type名MM来自于继承的单元)
# forcefield 和 By Type
write_once("Data Angles By Type"){
@angle: type @atom:atom1 @atom:atom2 @atom:atom3 @bond:b1 @bond:b2
}
2
3
这里介绍moltemplate最重要的功能之一。众所周知我们的逻辑结构不单单包括2-body的键接,还有3-body,4-body的angle和dihedral甚至还有improper。如果自己写程序去搜索拓扑结构的话会非常费事。因此这里给出了一个功能,只要你给出了结构的键接信息,那么就可以自动去查找所有的angle和dihedral和improper。其中语法也十分清晰明了,第一,指明这个angle的三个原子类型,第二,指明两个键接类型,仅此就可以遍历所有原子,标记出所有的角。注意命令名是Data Angles By Type。
# unit.lt
# -- example of a unit.lt inscript -- #
import "forcefield.lt"
Matrix inherits ForceField{
write("Data Atoms"){
# atom id mol id type charge coordinate
$atom:1 $mol:. @atom:M 0 1 1 1
$atom:2 $mol:. @atom:M 0 2 1 1
$atom:3 $mol:. @atom:M 0 3 1 1
$atom:4 $mol:. @atom:M 0 4 1 1
$atom:5 $mol:. @atom:M 0 5 1 1
}
write("Data Bonds"){
$bond:1 @bond:MM $atom:1 $atom:2
$bond:2 @bond:MM $atom:2 $atom:3
$bond:3 @bond:MM $atom:3 $atom:4
$bond:4 @bond:MM $atom:4 $atom:5
}
}
`Data Bonds`和`Data Bond list`的区别是什么。如果你使用了一个现成的力场`.lt`文件,请使用`Data Bond list`,可以根据指定的原子类型,自动赋予bond type。但是如果自己写力场文件,那么需要使用`Data Bonds`手动给出bond type。
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
# $和@计数器
如上面的$atom:1和@atom:1,都被称为变量。前面的符号是计数器,后面叫做类名(category),冒号后则是变量名,我们很快就会知道这个为什么要这么写。
计数器分为两种,$是动态计数器,每增加一个,在生成的时候都会赋给一个唯一的id,@是静态计数器,每一个类名在全局只会唯一给一个type。简单说,就是静态计数器和系统复杂度有关,动态计数器和系统规模有关。
# 变量作用域
上面提到的变量有两种形式,一种是全名,另一种是简名。简名通常是在一个结构对象(molecule-object)内,不会产生歧义。如果有多个分子,系统就不知道这个H是谁的,因此我们需要使用全名。
@cpath/catname:lpath
cpath:category的作用域,通常省略以表示全局。除了有极为特殊的要求之外不需要管
catname:类名
lpath:包括变量名和可选的指明那个分子的index
2
3
4
5
这样可以从一个结构对象内引用其他结构对象中的原子或者结构片段。但为了清晰起见,按照编程的基本原则,请不要交叉引用。
# $mol:. 和 $mol:...
你所看到的点,斜线的用法和unix操作系统文件路径表示方法一样,既可以用来表示相对路径,又可以表示绝对路径。所谓路径就是指的是不同的结构对象之间的关系(molecule-object)。类似的,一个点意为“这个”,就是代指本结构,如果一个结构单元中所有的原子都是$mol:.,那每new一次,同一个结构片段内mol id相同,不同结构片段mol id递增
What about $mol:...? I dont know. I just know one you want to build a macromolecule bottom up, to let the all units in macromolecue shall one mol id, then you can use $mol:.... And the most important is, you should add a create_var {$mol}
# system.lt
这一部分就是对之前已构建的所有单元片段的操纵了,不论是旋转,平移还是随机都是一行搞定。这些命令都在手册的一开头。
# 关于Debug
每次文件生成结束,都会给出一个名为output_ttree的文件夹,里面有各种文件。其中最重要的是就是ttree_assignments,这里面储存了计数器被替换的数和该计数器第一次出现的位置。